top of page


SULFORAFAN PROTECTS
AGAINST
CEREBRAL THROMBOTIC DYSFUNCTION
RESEARCH REPORT
British Journal of Pharmacology (2018) 175 3333–3346 3333
RESEARCH PAPER
The isothiocyanate sulforaphane modulates platelet function and protects against cerebral thrombotic dysfunction
Correspondence Felicity N E Gavins, Department of Molecular and Cellular Physiology, Louisiana State University Health Sciences Center Shreveport, 1501 Kings Highway, Shreveport, LA 71103, USA. E-mail: fgavin@lsuhsc.edu
Received 3 October 2017; Revised 30 April 2018; Accepted 4 May 2018
Scarlett Gillespie1,*, Paul M Holloway1,2,*, Felix Becker3 , Francesca Rauzi4 , Shantel A Vital2 , Kirk A Taylor4 , Karen Y Stokes2 , Michael Emerson4 and Felicity N E Gavins2,5
1 Division of Brain Sciences, Imperial College London, London, UK, 2 Department of Molecular and Cellular Physiology, Louisiana State University Health Sciences Center – Shreveport, Shreveport, LA, USA, 3 Department of General, Visceral and Transplant Surgery, University Hospital Muenster, Muenster, Germany, 4 Platelet Biology Group, National Heart and Lung Institute, Imperial College London, London, UK, and 5 Department of Neurology, Louisiana State University Health Sciences Center – Shreveport, Shreveport, LA, USA
*These authors contributed equally to this work.
BACKGROUND AND PURPOSE
Platelet activation provides a critical link between inflammation and thrombosis. Sulforaphane (SFN), a naturally occurring isothiocyanate, has been shown to display both anti-inflammatory and anti-thrombotic actions in the systemic microvasculature. As inflammation promotes thrombosis and vice versa, in this study we investigated whether SFN is able to reduce inflammatory potentiation of thrombotic events, suppress platelet activation and thrombus formation in the cerebral microvasculature.
EXPERIMENTAL APPROACH Thrombosis was induced in the murine brain using the light/dye-injury model, in conjunction with LPS treatment, with and without SFN treatment. In vitro and in vivo platelet assays (aggregation, flow and other functional tests) were also employed, using both human and murine platelets.
KEY RESULTS SFN was found to reduce LPS-mediated enhancement of thrombus formation in the cerebral microcirculation. In tail-bleed experiments, LPS treatment prolonged bleeding time, and SFN treatment was found to protect against this LPS-induced derangement of platelet function. SFN inhibited collagen-mediated platelet aggregation in vitro and in vivo and the associated adhesion and impaired calcium signalling. Furthermore, glycoprotein VI was shown to be involved in the protective effects observed with SFN treatment.
CONCLUSIONS AND IMPLICATIONS The data presented here provide evidence for the use of SFN in preventing stroke in selected high-risk patient cohorts. Abbreviations CRP, collagen-related peptide; CVX, convulxin; GPVI, glycoprotein VI; SFN, sulforaphane; vWF, von Willebrand factor
Introduction
Platelet activation and thrombus formation are essential mechanisms to limit blood loss at sites of vascular injury. However, inappropriate or excessive platelet activation has a detrimental role in the pathophysiology of numerous human disorders, including atherosclerosis (Aukrust et al., 2010), sepsis (Woth et al., 2011), sickle cell disease (Frelinger 3rd et al., 2014) and stroke (Fateh-Moghadam et al., 2007). Platelets may become activated and adhere to the damaged endothelium leading to vessel occlusion and ischaemic injury in downstream vascular beds. Enhanced platelet activation and thrombus formation are particularly relevant in stroke, which accounts for 129 000 annual deaths in the US alone (Mozaffarian et al., 2016). Inflammatory and coagulation pathways are intimately linked and have been demonstrated to cross-activate (Fiusa et al., 2015). LPS and inflammatory cytokines (e.g. TNF-α) increase platelet P-selectin and von Willebrand factor (vWF) on endothelial cells, thus enhancing platelet adhesion (Lou et al., 1997). The interdependence between inflammation and thrombosis is particularly evident in sepsis, where the systemic pro-inflammatory state favours a shift in the haemostatic balance towards a pro-coagulant state (Fiusa et al., 2015; Jung and Moroi, 1998). Systemic inflammation markedly increases platelet adhesion, fibrin deposition and propensity of platelet-dependent thrombosis within capillaries (Secor et al., 2010). Indeed, excessive platelet activation under inflammatory conditions can impair microvascular perfusion and propagate disseminated intravascular coagulation, tissue hypoxia and multiple organ dysfunction (Sakr et al., 2004). Sulforaphane (SFN), a naturally occurring isothiocyanate, can be found in cruciferous vegetables, for example, broccoli and cabbage (Bai et al., 2015). SFN has been demonstrated to have anti-inflammatory and anti-oxidative properties, exerting a beneficial modulation of NF-κB, activator protein-1 and initiation of anti-oxidative pathways (Zhang et al., 1992). ROS promote surface expression of P-selectin on platelets and endothelial cells enhancing platelet adhesion and coagulation. Thus, SFN may influence platelet activation and adhesion via both anti-inflammatory and anti-oxidative pathways. SFN inhibits human platelet aggregation in response to a number of receptor agonists by inhibiting PI3K/Akt pathways (Chuang et al., 2013). Furthermore, SFN treatment also reduces mortality in ADPinduced acute pulmonary thromboembolism (Jayakumar et al., 2013) and prolongs time to platelet plug formation in fluorescein sodium-induced platelet thrombi in murine mesenteric microvessels (Jayakumar et al., 2013). Despite these beneficial anti-inflammatory and anti-thrombotic features of SFN in the systemic microvasculature, its effect on thrombosis in the cerebral microcirculation is unknown. Therefore, we used in vitro and in vivo assays to investigate potential therapeutic benefits of SFN treatment in cerebral thrombosis in the context of inflammation and platelet activation.
Methods
Animals
Male mice aged 10–12 weeks were housed in controlledtemperature/humidity environment (22 ± 1°C, 60–70% relative humidity) in individual cages (five mice per cage, with wood shaving bedding and nesting material), with a 12 h light/dark cycle (lights on at 0700 h) and access to a standard chow pellet diet and tap water ad libitum. C57BL/6 mice were purchased from either Jackson Laboratory (Bar Harbor, ME, USA) or Harlan (Bicester, UK). Mice were allowed to acclimatize to their housing environment for at least 5 days prior to experimentation and to the experimental room for 1 h before experiments. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010a; McGrath and Lilley, 2015) and followed the European Union Directive (2010/63/EU) or were approved by the Louisiana State University Health Sciences Center Shreveport Institutional Animal Care and Use Committee and were in accordance with the guidelines of the American Physiological Society. All studies (animals and humans) were performed blinded and randomized, with a key system to identify which animal/sample had undergone which treatment. Furthermore, compounds administered were made by laboratory personnel other than the one performing the experiment. Animals were deeply anaesthetized, as determined by the absence of a pedal reflex, with ketamine (Ketaset, 150 mg·kg1 , Fort Dodge Animal Health, Southampton, UK) and xylazine (Rompun, 7.5 mg·kg1 , Bayer Healthcare, Newbury, UK) i.p. before being killed by cervical dislocation.
Photoactivation thrombosis model
Photoactivation of the cerebral microcirculation was performed in anaesthetized animals, as previously described (Gavins et al., 2011a,b). Animals were randomly assigned to treatment groups (n = 6 mice per group; Gavins et al., 2011a,b). SFN treatment or corn oil vehicle (Sigma-Aldrich) was administered i.p. 24 h prior to photoactivation. LPS (0.5 mg·kg1 ) or saline was administered 4 h prior to photoactivation. Briefly, a craniectomy was performed and artificial CSF (NaCl 131.9 mM, CaCl2 1.26 mM, CaCl2.2H2O 1.26 mM, KCl 2.95 mM, MgCl2.6H2O 0.64 mM, MgCl2 0.5 mM, (NH2)2CO 6.69 mM, C6H12O6 3.69 mM; Sigma-Aldrich) was placed on the cranial opening. The preparation was allowed to equilibrate for 30 min; 10 mg·kg1 of 5% FITC dextran (150 000 mol wt; Sigma-Aldrich) was injected i.v. and allowed to circulate for 10 min before photoactivation, which was initiated (excitation, 495 nm; emission, 519 nm) by exposing 100 μm of vessel length to epi-illumination with a 175-W xenon lamp (Lamda LS, Novato, CA, USA) and a fluorescein filter cube (HQ-FITC; Chroma, Bellows Falls, VT, USA) as described previously (Gavins et al., 2011a,b). Visualization of individual vessels and induction of thrombosis was performed using a 40× water-immersion objective attached to a Xanophot IVM microscope (HLX64610; Nikon, Melville, NY, USA). Epi-illumination was applied continuously, and time of blood flow cessation (≥60 s duration) was recorded in both venules and arterioles (30–70 μm). Epi-illumination was discontinued once blood flow ceased in the respective vessel. Video recordings were made using a silicon-intensified target camera (C-2400; Hamamatsu Photonics, Tokyo, Japan). Typically, two to four thrombi were induced per mouse and results averaged.
Analysis of bleeding time
Analysis of bleeding time in SFN, LPS or vehicle-treated animals was performed as previously described (Gavins et al., 2011a,b).
Platelet counts
Platelets from peripheral blood were stained with 3% citric acid and 10% crystal violet and manually counted using a haemocytometer.
Human platelet isolation
Written informed consent was obtained from human volunteers, and blood was collected with LSUHSC-S Institutional Review Board approval (IRB STUDY00000261) immediately prior to performing experiments using venipuncture of the cubital fossa. All experimental procedures were reviewed and approved by LSUHSC-S Institutional Review Board approval (IRB STUDY00000261). Volunteers were healthy, aged 18–45 and free from non-steroidal anti-inflammatory drugs 2 weeks prior to blood collection. Blood was collected with the anti-coagulant acidified sodium dextrose (ACD, 1:9; Sigma-Aldrich) and washed platelets isolated as previously described (Jones et al., 2010).
In vitro aggregometry
Aggregations were run with washed platelets for 3 min following addition of thrombin (Sigma-Aldrich), ADP (Sigma-Aldrich) or collagen (Pharmaceuticals International) as previously described (Jones et al., 2010).
Platelet velocity of aggregation [low angle light scattering (LaSca) method]
Human venous blood (5–6 mL) was obtained from healthy donors into an ACD (1:7) loaded 10 mL syringe. PRP was collected immediately by centrifugation of blood sample at 300 g for 5 min (RT). Platelet aggregation velocity of freshly isolated PRP (at final concentration of platelets ~1.25–1.67 × 107 cells·mL1 ) in a cuvette loaded with platelet media (NaCl 140 mM, KCl 2 mM, HEPES 10 mM, CaCl2 2 mM, glucose 5.5 mM, and MgCl 1 mM) was analysed using a laser particle analyser LaSca (Lumex, LTD, Saint-Petersburg, Russia) as described previously (Gavins et al., 2011b). Once a constant basal signal of platelet suspension was established (1–2 min), platelet aggregation was induced with convulxin (CVX; 1.7 ng·mL1 ), LPS (7.5 μg·mL1 ; 5 min incubation prior to CVX exposure) or a combination of CVX and LPS (1.7 ng·mL1 and 7.5 μg·mL1 respectively). Several samples of PRP were also preincubated with vehicle or SFN (60 μM) 30 min at RT prior to platelet aggregation being induced with CVX, LPS or LPS and CVX. No platelet preparation was exposed to more than one concentration of either CVX or LPS at any one time. The velocity of platelet aggregation was analysed by the LaScA method, and a normalized velocity of platelet aggregation was calculated using original software LaSca_32 as previously described (Gavins et al., 2011b).
In vivo aggregation
We previously developed and characterized a model for measuring thromboembolic platelet aggregation in vivo (Tymvios et al., 2008). This model was developed as a refined model of thromboembolic models that use death as an endpoint (DiMinno and Silver, 1983). Blood was collected from terminally anaesthetized donor mice by cardiac puncture and platelets isolated and radiolabelled with 1.8 MBq of indium111 oxine (Tymvios et al., 2008). Radiolabelled platelets were infused via the femoral vein into anaesthetized (urethane 25 % w. v-1, 10 μL·g1 ) recipient mice. Platelet aggregation was measured as increase in platelet-associated counts in the pulmonary vasculature via a 1 cm diameter scintillation probe over the thoracic region following i.v. injection of non-lethal concentrations of collagen (50 μg·kg1 ). Vehicle or SFN (10 mg·kg1 ) was administered i.p. 30 min before collagen.
Ca2+ signalling
Platelets from PRP were incubated at 37°C with CaCl2 (200 μM) and the ionophore Fura2-AM (5 μM; Sigma-Aldrich) for 30 min in the dark. The PRP was then supplemented with 5 μg·mL1 PGE1 and 12 μL·mL1 ACD, and centrifuged (400× g, 10 min). Platelets were resuspended in THB+ at 2.5 × 108 cells·mL1 and treated with SFN (1–100 μM) for 2 min prior to measurement of fluorescence at excitation wavelengths of 340 and 380 nm, and an emission wavelength of 500 nm using a Biotech Synergy H1 hybrid plate reader (BioTek, Winooski, VT, USA) at 37°C with constant agitation. Platelet lactate dehydrogenase (LDH) assay LDH release was detected as a measure of cytotoxicity in washed platelets (5 × 107 cells·mL1 ) in response to varying concentrations of SFN, using the Roche cytotoxicity detection kitPLUS (Roche, Nutley, NJ, USA) as per the manufacturer’s instructions. Absorbance was read at 490 nm using a Biotech Synergy H1 hybrid plate reader (BioTek).
Platelet-collagen adhesion under flow
Washed platelets were labelled with 5 μM carboxyfluorescein succinimidyl ester (Affymetrix, CA, USA) for 30 min. Platelets were centrifuged (800× g) and resuspended in Tyrode’s solution with 3 mg·mL1 BSA and 5 mM glucose to 2.5 × 107 cells·mL1 and mixed with erythrocytes to give a haematocrit of 40%. Cells were then perfused over collagen coated IBIDI μ-Slide VI0.4 flow chambers (IBIDI, WI, USA). To investigate platelet adhesion to collagen under venular shear rates, platelets were perfused at 1 dyne·cm2 for 10 min. For investigation of vWF-mediated collagen adhesion under high shear rates, IBIDI μ-Slide VI0.4 flow chambers were pre-coated with vWF (2 μg·mL1 ) in Tyrode’s buffer + BSA and glucose, and platelets were perfused at 5 dyne·cm2 (0.5 Pascal) for 4 min followed by perfusion of Tyrode’s buffer alone for 1 min. Platelets were perfused over vWF coated surface at 5 dyne·cm2 for 5 min. Buffer alone was then perfused over the surface for 1 min before images were captured using an Olympus IX71 inverted microscope (Olympus, Saucon, PA, USA) with an Olympus DP70 camera and analysed with ImageJ (NIH) software. Data are presented as % surface area covered. SFN treatments were given 30 min before flow.
Murine platelet flow cytometry
JON/A-PE and P-selectin FITC antibodies (Emfret Analytics, Eibelstadt, Germany) were used to measure collagen-related peptide (CRP)-induced activation of integrin αIIbβ3 receptors and surface exposure of P-selectin in murine platelets using flow cytometry, as previously described (Yan et al., 2013). IgG isotype antibodies were used as controls. A platelet pellet was obtained from PRP and resuspended in Tyrode’s solution. The platelets were treated with Fc block (15 min, RT) followed by SFN (60 μM) or vehicle for 30 min. The antibodies (1:8 dilution) were added before CRP (10- μg·mL1 , Collagen Toolkits, Cambridge, UK) was used to stimulate the platelets for 15 min. The activation was stopped by the addition of 450 μL of 1% paraformaldehyde. Platelets were identified by their light scattering using an LSRII flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) before two-colour analysis of at least 10 000 events per sample was performed.
Human platelet flow cytometry
AlexaFluor647 conjugated anti-human CD41/CD61 antibody (BioLegend; clone PAC-1; Cat #362806) and Alexa Fluor 647 mouse IgMk Isotype control (BioLegend; clone MM-30; Cat #401618) were used in 1:100 dilution to determine % of integrin αIIbβ3 complex positive platelets. FITC conjugated anti-human CD62P (P-selectin) (Emfret; clone AK4; Cat# 304904) and FITC mouse IgG1k isotype control (Emfret; clone MOPC-21; Cat #400107) in 1:100 dilution was used to established P-selectin positive platelets. Human venous blood (5–6 mL) was obtained from healthy donors into an ACD (1:7) loaded syringe. PRP was collected immediately by centrifugation of blood sample at 300× g for 5 min (RT). The topical layer of PRP was transferred into new eppendorf tubes and centrifuged at 750× g for 10 min (Thermo Scientific Legend Micro 17 centrifuge). Supernatant was discarded, and platelet pellet was reconstituted with Tyrode’s solution (no Ca2+). Fc block was used to inhibit non-specific binding according to the manufacturer’s instruction (eBioscience, San Diego, CA, USA). Platelets were reconstituted with Tyrode’s solution with 1 mM Ca2+ at final concentration 1 × 106 cells. 25 μL-1 and incubated with SFN (60 μM) for 30 min, LPS (7.5 μg·mL1 ) for 5 min, and with a combination of SFN and LPS, then stained with antibodies and isotopes. Selected platelet samples were stimulated with CVX (1.7 ng) for 15 min at 37°C, and platelet reactivity was stopped by adding 1% formaldehyde. The platelet samples were assayed using a flow cytometry assay with BD LSRII SORP immediately after fixation. A total of 20 000 events were recorded per sample and analysed by DivaSoftware 8.0.1.
Data analysis and statistical procedures
All data were analysed using GraphPad Prism 6 for MacOS X (Version 6). Data are expressed as mean ± SEM with n values given in the respective figure legends. When determining statistical significance between two groups, an unpaired t-test was carried out and where appropriate, corrected for multiple comparisons using the Holm–Šídák method. Multiple groups were analysed by one-way ANOVA or non-parametric Kruskal–Wallis test and post hoc comparisons were performed by a Bonferroni or Dunn’s multiple comparison test respectively. In vivo platelet aggregation data are expressed as the percentage increase in maximal radioactive counts from the baseline recordings or AUC. A Student’s t-test was used to compare mean values. Differences were considered statistically significant if P < 0.05. The figures have been graphically presented on a range-specific axis. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018).
Materials
The following drugs were used at the most effective doses/concentrations based on our previous findings and dissolved in sterile saline unless otherwise stated: SFN treatment or corn oil vehicle (Sigma-Aldrich St Louis, MO, USA); LPS (0.5 mg·kg1 , Sigma-Aldrich); thrombin (Sigma-Aldrich); ADP (Sigma-Aldrich); collagen (Pharmaceuticals International, Linz, Austria) and convulxin (CVX, Cayman Chemical Company, MI, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology. org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Results
SFN treatment inhibits thrombus formation in
the inflamed cerebral microvasculature
Thrombus formation, assessed as time to blood flow cessation in murine cerebral arterioles and venules, was quantified using the light dye injury model and intravital fluorescence microscopy (Figure 1A–C). Time required for complete flow cessation was longer in arterioles than venules, as reported previously (Gavins et al., 2011a,b; Yan et al., 2013). LPS (0.5 mg·kg1 , 4 h) reduced time to flow cessation, demonstrating the pro-thrombotic environment within the inflamed cerebral microvasculature following systemic LPS exposure (Figure 1). We then assessed the influence of SFN pretreatment on this accelerated thrombus formation. SFN treatment was found to have no effect on thrombus formation under non-inflammatory conditions. However, SFN (5 and 50 mg·kg1 administered 24 h prior to saline or LPS treatment) reversed the LPS-induced reduction in thrombotic time in both cerebral venules and arterioles (Figure 1).
SFN treatment reverses LPS-induced
prolongation of bleeding in vivo
In order to investigate whether SFN could also influence coagulation following peripheral injury, tail-bleeding experiments were performed. LPS treatment prolonged tailbleeding time and reduced the number of circulating platelets (Figure 2A, B). Whilst in the absence of inflammatory challenge SFN treatment had no effect on tail-bleeding time, in LPS treated animals, SFN was found to counteract the LPS-induced delay in cessation time (Figure 2A). No significant effect was observed in circulating platelet number in LPS-treated animals with and without SFN (Figure 2B).
SFN treatment directly inhibits human platelet
aggregation
Having implicated a protective role for SFN in reversing the LPS-induced pro-thrombotic cerebral microvascular

Figure 1
SFN treatment inhibits thrombosis formation in the inflamed cerebral microcirculation. Light/dye-induced thrombosis was visualized in the brain and quantified in terms of time taken for complete blood flow cessation. Images taken of thrombus formation in the brain arterioles during light/ dye thrombosis events at (A) 0 min, (B) 21 min and (C) 29 min when blood flow cessation occurred. Values represent time to flow cessation following light/dye injury in cerebral (D) arterioles and (E) venules. Animals were treated with SFN (5 or 50 mg·kg1 ) or vehicle 24 h prior to inflammatory challenge with LPS (0.5 mg·kg1 ). Bar = 10 μm. Data are mean ± SEM of 6 mice per group with one to three vessels analysed per mouse. * P < 0.05 versus vehicle control. + P < 0.05 versus LPS treated group.

Figure 2
SFN treatment inhibits the LPS-induced prolonged bleeding time. (A) Duration of tail-bleeding time and (B) peripheral blood platelet counts following SFN (50 mg·kg1 ) or vehicle (saline) 24 h prior to inflammatory challenge with LPS (0.5 mg·kg1 ). Data are mean ± SEM of 5 mice per group. * P < 0.05 versus corresponding vehicle group. + P < 0.05 versus LPS treated group.
phenotype in vivo, we next examined whether SFN may directly act on platelets to inhibit aggregation and thrombus formation in response to thrombin, ADP or collagen. Light transmission aggregometry experiments were performed on isolated human platelets. SFN (0.1–100 μM) did not induce any platelet aggregation above vehicle alone, whilst thrombin (0.1 U·mL1 ) demonstrated a robust response (Figure 3A). When SFN treatments were given 2 min prior to activation with thrombin, no influence over platelet aggregation was observed (data not shown). However, if SFN was administered 30 min prior to activation, only the highest concentration (100 μM) tested was found to induce a significant reduction of platelet aggregation (Figure 3B). When given 2 min prior to activation, the highest concentration
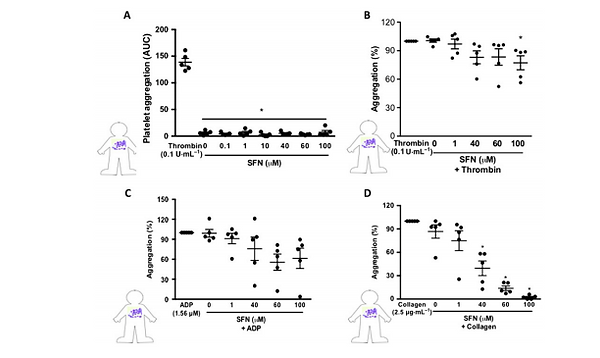
Figure 3
SFN treatment reduces human platelet aggregation in vitro. Light transmission aggregometry was used to investigate the influence of SFN treatment (0.1–100 μM) for 30 min on isolated platelet aggregation responses to a number of platelet agonists: (A) no agonist, (B) thrombin, (C) ADP and (D) collagen. All aggregations were run for 3 min following addition of the platelet agonists. Aggregation responses are expressed as a percentage of aggregation induced by platelet agonist alone. Data are mean ± SEM of n = 5 independent donors per group. * P < 0.05 versus control.
of SFN tested was able to inhibit platelet aggregation in response to ADP stimulation (data not shown); however, all lower concentrations were ineffective. No effect was observed when SFN was administered 30 min prior to ADP stimulation (Figure 3C). SFN treatment did however show a robust inhibition of collagen-stimulated platelet aggregation, with both 60 and 100 μM significantly reducing collagen responses when given 2 min prior (data not shown). When given 30 min before activation, all but the lowest concentration of SFN were found to provide a significant inhibition of platelet aggregation in response to collagen (Figure 3D). Indeed, 60 and 100 μM treatments were found to completely abolish platelet collagen responses.
SFN treatment inhibits collagen-induced
platelet aggregation in vivo
Having shown an effect of SFN on isolated human platelet aggregation, we explored the relevance of this in vivo using an established mouse model of radiolabelled platelet aggregation. Collagen (50 μg·kg1 i.v.) induced a transient increase in platelet-associated counts that returned to baseline (Figure 4). This has previously been shown to represent reversible aggregation of platelets (Jones et al., 2010). Treatment with SFN (10 mg·kg1 ) for 30 min before injection of collagen had no effect on the amplitude of platelet aggregation response but significantly reduced the duration of the response measured as AUC (Figure 4).
SFN suppresses collagen induced Ca2+ signalling
Platelet intracellular calcium signalling is crucial for platelet aggregation and thrombus formation and is activated upon vWF and collagen binding. Thus, effects of SFN on calcium signalling were investigated. Consistent with the lack of aggregatory responses to SFN, treatment with SFN alone did not induce calcium signalling in isolated human platelets (Figure 5A). Collagen stimulation of human platelets was found to induce significant transient increases in intracellular calcium levels as detected by an increased ratio of 340/380 nm excitation emission of Fura 2AM dye.

Figure 4
SFN reduces collagen-induced platelet aggregation in vivo. A thromboembolic in vivo model was used to measure platelet aggregation in mice treated with SFN (10 mg·kg1 ) or vehicle saline (V) for 30 min prior to an i.v. injection of collagen (50 μg·kg1 ). Platelet aggregation was measured by changes in platelet-associated radioactivity (10 min), and data are represented as (A) maximum % increase in platelet counts, (B) AUC and (C) % of change in platelet counts. Data are mean ± SEM of n = 5 mice per group; * P < 0.05 versus saline vehicle treated group. + P < 0.05 versus collagen stimulated vehicle group.
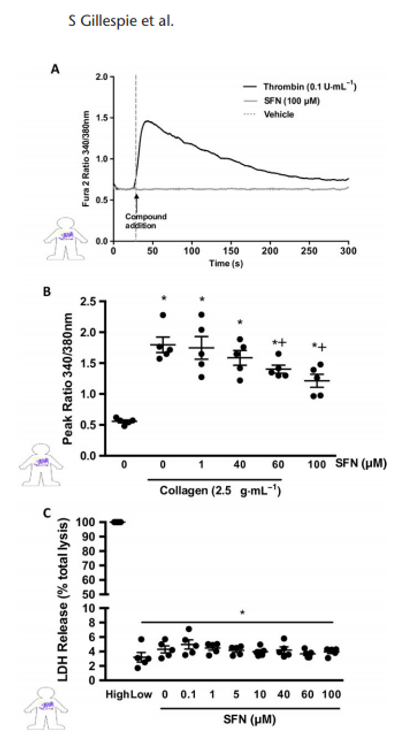
Figure 5
SFN influence over collagen-induced Ca2+ signalling. Cytosolic calcium release was measured using Fura 2AM dye in human platelets. (A) Representative trace showing temporal increase in Fura 2 AM fluorescence following administration of the positive control (0.1 U·mL1 thrombin) but with no effect of SFN treatments alone. (B) Histogram of peak 340/ 380 nm emission, in platelets stimulated with 2.5 μg·mL1 collagen following SFN treatment. (C) LDL release as a marker for cytotoxicity following SFN treatment, data presented as a % LDH release when compared to completely lysed cells. Data are mean ± SEM of n = 5 independent donors per group. * P < 0.05 versus un-stimulated control. + P < 0.05 versus collagen stimulated vehicle group.
Treatments with 60 and 100 μM SFN, 2 min prior to stimulation, were found to significantly inhibit collagen-induced intracellular calcium signalling events (Figure 5B). In order to test ensure the observed reduction in platelet responses was not due to reduced cell viability at high SFN concentrations, an LDH assay was performed. Across all treatments tested, cells remained intact (Figure 5C).
SFN inhibits platelet adhesion under
physiological flow
The above experiments demonstrate that SFN strongly inhibits collagen-induced platelet–platelet aggregation. In vivo, at sites of vascular injury, platelets come into contact with sub-endothelial collagen allowing them to adhere, triggering their activation and inducing subsequent thrombus formation. Thus, it is important to investigate levels of platelet adhesion to a collagen substrate under physiological flow conditions. CSFE labelled human platelets were perfused at shear rates physiologically relevant to venular shear stress, and platelet adhesion was monitored. SFN treatment (100 μM) 30 min prior to flow was found to significantly reduce platelet adhesion to collagen, as represented by a reduced platelet coverage of collagen coated surface following 10 min of flow (Figure 6A, B).
Whilst under static conditions and at low shear rates platelets may directly adhere to collagen, in conditions where shear stress is above 5–10 dynes· cm2 (0.5-1 Pascals) vWF is a crucial determinant of thrombus formation. Circulating vWF is activated by immobilization at sites of vascular injury and the action of shear stress exposes binding sites in the vWF-A1 domain, allowing interaction with platelet GPIbα. As such, arteriole thrombosis in humans requires vWF interaction. To investigate vWF-mediated platelet adhesion to collagen, isolated human platelets were perfused over vWF-activated collagen IV at a shear stress of 5 dynes·cm2 (0.5 Pascal), and platelet adhesion was assessed. Unlike at the lower shear of 1 dyne·cm2 , in the absence of vWF, very few platelets were found to adhere to collagen at 5 dyne·cm2 within the time frame tested. However, perfusion of vWF for 4 min prior to the introduction of platelets resulted in a greater than 20-fold increase in platelet adhesion. Platelet pretreatment with 1, 40 and 100 μM (Figure 6C) SFN 30 min prior to flow was found to reduce the platelet-vWF-collagen adhesion (Figure 6D).
Effect of SFN is via a suppression of the
glycoprotein VI (GPVI) pathway in murine platelets
In light of the observed effects of SFN on collagen in vivo and in vitro under physiological flow conditions, and the fact that, in addition to vWF-mediated platelet–collagen interactions, GPVI is a platelet receptor crucial to the collagen-induced activation and aggregation, flow cytometry of isolated murine platelets was performed to investigate the influence of SFN on GPVI-mediated platelet activation. Activation status was measured by expression of the activated form of the integrin αIIbβ3 receptor using the JON/A antibody (DiMinno and Silver, 1983) and surface expression of P-selectin (Figure 7). Whilst the GPVI-selective peptide CRP stimulated platelets to increase levels of activated αIIbβ3 receptors, SFN treatment (60 μM) was found to suppress these levels (Figure 7A, C). This anti-activation effect of SFN was further evidenced by the SFN-mediated reduction of CRP-induced P-selectin expression (Figure 7B, D).
SFN is via a suppression of the GPVI pathway
in LPS-induced inflammatory setting
Next, we wanted to translate these findings to the clinical setting and further tease out the mechanism of action of SFN in

Figure 6
SFN treatment reduces collagen and vWF mediated collagen platelet adhesion under conditions of physiological flow. (A and B) Human platelets were perfused over collagen-coated flow chambers at a shear stress of 1 dyne·cm2 (0.1 Pascal) for 10 min before imaging, SFN treatments were given 30 min prior to flow. (C and D) Collagen coated flow chambers were pretreated with vWF (2 μg·mL1 ) before perfusion of platelets at a shear stress of 5 dyne·cm2 (0.5 Pascal), and SFN given 30 min prior to flow. Data are mean ± SEM of n = 5 independent donors per group. * P < 0.05 versus vehicle treated in the absence of vWF. + P < 0.05 versus vWF treated vehicle group.
an inflammatory backdrop (i.e. LPS). We assessed the effect of SFN on platelet P-selectin and αIIbβ3 receptor expression following stimulation with the GPVI collagen receptor agonist CVX. Figure 8 shows that CVX induced a marked increase in platelet activation (Figure 8A, B), which was suppressed by co-administration of SFN (60 μM). Stimulation with LPS (7.5 μg·mL1 ) alone did not affect platelet αIIbβ3 or P-selectin levels but it potentiated these markers of platelet activation when in the presence of CVX (which concurs with Lopes Pires et al., 2017). SFN treatment (60 μM) was found to suppress these effects (Figure 8C, D), suggesting that SFN reduces platelet activation under inflammatory conditions via GPVI. To analyse the effects of LPS on platelet aggregation velocity, PRP was incubated with LPS (0, 1 and 7.5 μg·mL1 ; Lopes Pires et al., 2017) in the presence or absence of CVX. Whilst no effect of LPS alone was observed with platelet aggregation velocity (Supporting Information Figure S1), in the presence of CVX, accelerated aggregation was observed (which concurs with previous findings (Lopes Pires et al., 2017). Furthermore, these effects were reduced by the coadministration with SFN (Figure 8C, D), suggesting that SFN also reduces the velocity at which platelets aggregate in inflammatory conditions by interfering with GPVI signalling.
Discussion
To evaluate the potential of SFN to reduce platelet thrombotic function during inflammation, we utilized two distinct in vivo models: tail-bleeding time and cerebral intravascular occlusion in combination with LPS treatment to study the inflammatory perturbations of platelet activity. Consistent with the observation of sepsis-induced thrombocytopenia frequently observed in humans, we found LPS treatment in mice reduces the number of circulating platelets. In tail-bleeding experiments, LPS administration was found to prolong bleeding time, whilst in the cerebral microcirculation, light/dyeinduced thrombosis time was significantly reduced in LPS
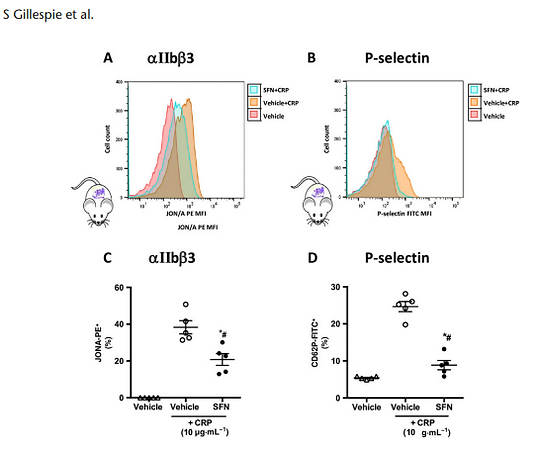
Figure 7
Platelet expression of cell surface receptors following CRP stimulation. Activated integrin αIIbβ3 and P-selectin were quantified using flow cytometry in platelets following 15 min stimulation with CRP at 10 μg·mL1 . Vehicle or SFN treatment (60 μM) was given 30 min prior to activation. (A and B) representative histograms of activated αIIb β3 and P-selectin expression on platelets treated with SFN/vehicle +/ CRP stimulation. Mean fluorescence intensities of platelet (C) activated integrin αIIbβ3 (GPIIbIIIa) and (D) P-selectin. Data are mean ± SEM of n = 5 mice per group. * P < 0.05 versus control (vehicle) and CRP group. # P < 0.05 versus CRP only control group.
treated animals. These apparently contradictory findings are also evident in sepsis, which is associated with an initial activation of coagulation and fibrinolysis with subsequent depletion of anticoagulant systems via endothelial disruption and glycocalyx degradation releasing heparin-like substances (Burgess et al., 2000; Patel et al., 2008; Youn et al., 2010). As such sepsis-induced disseminated intravascular coagulation is characterized by simultaneously occurring thrombotic and bleeding problems (Gavins et al., 2011b), as has been observed here in LPS stimulated mice. SFN treatment was found to counteract enhanced platelet activation and thrombus formation in the cerebral microcirculation by prolonging time to blood flow cessation. However, opposite effects were observed in the tail-bleeding model, in which SFN expedited time to cessation of bleeding. The bleeding assay is widely used as an in vivo assessment of haemostatic action of platelets (Zhang et al., 2009); hence, the opposing effects are likely due to the mechanisms regulating these two processes of thrombosis and haemostasis not being identical and effects being quantified outside (tailbleeding model) and inside the body (thrombosis model (Liu et al., 2012). Clinically, this is an important finding in that SFN protected against the pro-thrombotic disorder not only without increasing bleeding risk in our model but actually alongside reducing bleeding. The influence of LPS on thrombotic function likely occurs through a number of mechanisms, for example, the direct influence of LPS on platelet function has been demonstrated through the activation of platelet cell surface TLR4, inducing platelet activation and secretion of dense and alpha granules and hence ATP release and P-selectin expression (Zhang et al., 2009). The ability of SFN to suppress TLR4 oligomerization in a thiol-dependent manner (Youn et al., 2010), along with other SFN anti-inflammatory mechanisms, may explain the lack of SFN’s influence over in vivo thrombosis in the absence of pathological LPS challenge. In addition, some redundancy has previously been demonstrated in the tail-bleed model between thrombin and collagen mediated platelet activation (Bynagari-Settipalli et al., 2014). Ablation of collagen signalling had previously been shown to have no effect on tail bleeding if thrombin signalling remains intact (BynagariSettipalli et al., 2014). Whilst in vitro we observed a robust inhibition of collagen induced aggregation with reduced intracellular signalling and collagen adhesion, relatively little effect was observed upon thrombin-induced activation. This preservation of thrombin signalling may explain the observed differences in SFN effects between the two models utilized.

Figure 8
Human platelet expression of cell surface receptors post CVX stimulation. Activated integrin αIIbβ3 and P-selectin were quantified using flow cytometry in platelets following 15 min stimulation with the GPVI collagen receptor agonist CVX (1.7 ng). Vehicle or SFN treatment (60 μM) was given 30 min prior to activation, followed by vehicle (saline) or LPS (7.5 μg·mL1 for 5 min). Mean fluorescence intensities of platelet (A and C) activated αIIbβ3 (GPIIbIIIa) and (B and D) P-selectin. (E) Velocity of aggregate formation was measured using low angle light scattering technique for platelets stimulated by CVX, CVX + LPS or CVX + LPS + SFN (concentrations as above). Data are mean ± SEM of n = 5–6 mice per group [n = 4 for vehicle (P-selectin) only and LPS (P-selectin) only groups due to an outlier in each]. # P < 0.05 versus control (vehicle) only group. $ P < 0.05 versus LPS only group. **P < 0.05 versus control (vehicle) and CVX group. @P < 0.05 versus LPS and CVX group.
In order to further study effects of SFN treatment on platelet function, we performed a number of in vitro studies using human platelets. We found SFN to have a small influence over ADP and thrombin-mediated platelet aggregation but found SFN to be most effective in reducing collagen-induced platelet activation. The observation that SFN has potent inhibitory actions on platelet aggregation is not unique to this study and has previously been demonstrated by others using in vitro collagen-induced aggregation (Kilkenny et al., 2010b; Jayakumar et al., 2013) and collagen-induced thromboembolic mortality in vivo (Kilkenny et al., 2010b). Thus, in order to further investigate this phenomenon, effects of SFN on collagen-mediated calcium signalling and platelet adhesive activity to collagen under physiological flow conditions were investigated. SFN treatment was found not only to reduce in vitro aggregation but also to inhibit both collagen-induced intracellular signalling and platelet binding/adhesion to collagen, suggesting SFN may interfere with collagen receptor binding. In vivo studies confirmed that the ability of SFN to inhibit collagen-induced platelet aggregation was relevant in an in vivo setting where platelets circulate in whole blood under the influence of negative mediators of platelet activation generated by the vascular endothelium. Therefore, effects of SFN on platelet aggregation were not an artifactual observation occurring only in isolated platelets. In contrast to earlier studies using thromboembolic mortality, effects of SFN here can conclusively be attributed to an effect on platelets rather than non-specific factors involved in the death of the animal. Considering collagen-induced platelet aggregation has been demonstrated to be dependent on ADP (Gavins et al., 2011a) and that in the present study SFN treatment profoundly influenced platelet collagen responses but had little effect on ADP-induced platelet activation, these results suggest interference in platelet collagen signalling upstream of ADP, perhaps at the receptor level. Whilst platelet adhesion to collagen is evident under static conditions or at low shear rates platelets, under levels of higher shear rates, relevant to conditions in arterioles vWF is crucial for platelet adhesion and thrombus formation. Thus, experiments were performed to assess the extent at which SFN may influence collagen–vWF–platelet interactions, finding SFN treatment to also inhibit vWF-mediated adhesion. Both collagen activation and vWF binding of platelets are highly relevant to the pathology of atherosclerosis and stroke, where extracellular matrix exposure leads to platelet collagen binding via vWF. Elevated circulating levels of vWF have been demonstrated to remain elevated for up to 3 months following stroke (Tang et al., 2014). The initial platelet–vWF–collagen interaction is via platelet GPIba and tethers the platelets to the endothelium. However, immobilization and subsequent activation of platelets requires direct binding of platelet GPVI to collagen, as evidenced by the failure of platelets from GPVI-deficient mice to respond to collagen (DiMinno and Silver, 1983). GPVI, via inside-out signalling, enhances the affinity of other integrins such as αIIbβ3, ultimately leading to platelet adhesion. As such, GPVI plays a central role in thrombosis. Here, flow cytometry was utilized to investigate whether the antithrombotic effects observed with SFN was by interfering with GPVI signalling. By directly stimulating the GPVI pathway with CRP, we found that SFN was able to decrease activation of αIIbβ3 receptors and surface expression of P-selectin. Thus, GPVI is involved in the protective effects observed with SFN treatment. It is interesting to note that SFN has already been previously shown to significantly inhibit platelet–neutrophil aggregation (along with P-selectin and GPIIb–IIIa expression) in subjects with metabolic syndrome versus healthy controls (Konić-Ristić et al., 2013). With respect to our study, whilst the effects of SFN on the interactions between leukocytes and platelets are not the main aim of our study, we found that SFN reduces expression of GPIIbIIIa and Pselectin on platelets which may reduce platelet–leukocyte interactions through P-selectin inhibition by increasing levels of cAMP followed by inhibiting intracellular signals (such as the PI3K/Akt and PLCγ2-PKC-p47 cascades) and ultimately inhibiting platelet activation. Additionally, we found these effects of SFN on the markers of platelet activation (i.e. GPIIbIIIa and P-selectin) and furthermore also on platelet aggregation velocity to also hold true when platelets were assessed in vitro under inflammatory conditions. Our findings suggest that SFN is able to shift the haemostatic balance towards an anti-coagulant state. Here, we have shown that SFN reduces LPS-mediated light/dye-induced thrombus formation within the cerebral microvasculature in vivo. LPS effects the expression of GPVI, GP1b (vWF receptor) (Liu et al., 2013) and integrin α2β1 (type 1 collagen receptor) on platelets and collagen and vWF on endothelial cells (Echeverría et al., 2013). Both collagen and vWF activate platelet GPVI and GP1b, stimulating Ca2+ signalling in platelets and resulting in the overexpression of GPIIb/IIIa and integrin α2β1 on the surface of the platelet. These effects lead to accelerated platelet adhesion, activation, aggregation and exacerbated thrombus formation. Our findings show that the effect of SFN on LPS-mediated thrombotic responses may be dependent on the involvement of GPVI receptor on platelets as well as collagen. SFN may also inhibit LPS-enhanced thrombosis by decreasing Ca2+ mobilization which may affect platelet activity, at least in part, through adenylate cyclase (AC)/cAMP followed by inhibiting intracellular signals (such as the PI3K/Akt and PLCγ2-PKC-p47 cascades) and ultimately inhibiting platelet activation (Jayakumar et al., 2013). In addition, as SFN reduced thrombin-induced aggregation, it is highly likely that the effects of SFN are mediated by protease-activated receptors PAR3 and PAR4, but not through ADP-receptors (P2Y1 and P2Y12) (as demonstrated by a lack of response of SFN against ADP-induced aggregation). Thus, inhibition of the LPS-induced thrombotic responses by SFN in combination with the effectiveness of SFN in suppressing collagen-induced Ca2+ mobilization, platelet adhesion underflow (GPVI) and platelet GPIIb/IIIa expression demonstrates the effectiveness of SFN in GPVI-mediated signalling as a potential therapeutic target and anti-thrombotic strategy. In nucleated cells, SFN activates the transcription factor Nrf2 through binding cystine residues on KEAP1, although SFN has been shown to promiscuously bind to cysteine on thiol groups and thus influence protein function (Zhang et al., 1992). GPVI activation has been shown to involve receptor oligermization (Yan et al., 2013) stabilized through disulfide bonding between cysteinyl thiol groups (Yan et al., 2013). Thus, considering the prevalent role of SFN in binding thiol groups, it cannot be ruled out that SFN inhibits receptor dimerization through interaction with cystinyl-thiol groups. Furthermore, SFN has been previously shown to suppress oligomerization of TLR4 in a thiol-dependent manner, as mentioned above. Platelet thiol-disulfide balance is highly influential in regulating platelet function and upon activation 440% increase in surface protein thiol groups has been observed (Youn et al., 2010). Conformational changes in GPIb upon platelet activation have been shown to result in exposure of free thiols (Liu et al., 2012). Moreover, thiol groups are important in a number of other integrins such as αIIbβ3, which may indirectly interact with collagen via vWF. Thus, interaction with GPVI, as indicated by our experimental results, may occur through thiol groups. However, further investigations are needed to determine whether SFN modifies extra and/or intracellular thiol groups on GPVI. The efficacy of anti-platelet agents is often limited by bleeding complications. Thus, the present finding that SFN inhibited both, simultaneously, the LPS-induced elevated peripheral bleeding and increased propensity for intravascular thrombosis, whilst having no effect in saline treated animals is most promising for the treatment of thrombotic disorders and inflammatory related thrombotic complications. The present study demonstrates the protective functions of SFN pretreatment in the brain by normalizing platelet thrombotic functions under inflammatory conditions, which is relevant to prophylactic strategies. The therapeutic benefit of SFN as an acute treatment to inhibit platelet activation, subsequent to thrombotic or inflammatory events, will require further investigation. However, the present findings are promising, in that SFN provides a modulation of platelet activation in the context of a pathological inflammatory environment rather than an indiscriminant inhibition of platelet activation, thus potentially avoiding the bleeding complications associated with current anti-thrombotic medications.
Acknowledgements
The authors thank Antonia Solomon, Platelet Biology Group, National Heart and Lung Institute, Imperial College London and also the LSUHSC-S core facility. F.N.E. Gavins is supported by the National Institutes of Health/National Heart, Lung, and Blood Institute [NIH/NHLBI (HL125572-01A1)]. F. Rauzi and M. Emerson are supported by a project grant from National Centre for the Replacement, Refinement and Reduction of Animals in Research (NC/M000079/1). K. Stokes is supported by American Heart Association grant in aid: 15GRNT25910012.
Author contributions
S.G., P.M.H., S.A.V., F.R., K.A.T., K.Y.S., M.E. and F.N.E.G. designed the experiments. S.G., P.M.H., S.A.V., F.R., K.A.T. and F.N.E.G. performed the experiments. F.B., K.Y.S., M.E. and F.N.E.G. provided scientific input. S.G., P.M.H., S.A.V., F.R., K.A.T., F.B., M.E. and F.N.E.G. interpreted the results. All authors helped write manuscript and reviewed the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and
scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
References
Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al. (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271.
Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al. (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359.
Aukrust P, Halvorsen B, Ueland T, Michelsen AE, Skjelland M, Gullestad L et al. (2010). Activated platelets and atherosclerosis. Expert Rev Cardiovasc Ther 8: 1297–1307.
Bai Y, Wang X, Zhao S, Ma C, Cui J, Zheng Y (2015). Sulforaphane protects against cardiovascular disease via Nrf2 activation. Oxid Med Cell Longev 2: 9–14.
Burgess JK, Hotchkiss KA, Suter C, Dudman NP, Szollosi J, Chesterman CN (2000). Physical proximity and functional association of glycoprotein 1balpha and protein-disulfide isomerase on the platelet plasma membrane. J Biol Chem 275: 9758–9766.
Bynagari-Settipalli YS, Cornelissen I, Palmer D, Duong D, Concengco C, Ware J (2014). Redundancy and interaction of thrombin- and collagen-mediated platelet activation in tail bleeding and carotid thrombosis in mice. Arterioscler Thromb Vasc Biol 34: 2563–2569.
Chuang WY, Kung PH, Kuo CY, Wu CC (2013). Sulforaphane prevents human platelet aggregation through inhibiting the phosphatidylinositol 3-kinase/Akt pathway. Thromb Haemost 109: 1120–1130.
Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA et al. (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Br J Pharmacol 175: 987–993.
DiMinno G, Silver MJ (1983). Mouse antithrombotic assay: a simple method for the evaluation of antithrombotic agents in vivo. Potentiation of antithrombotic activity by ethyl alcohol. J Pharmacol Exp Ther 225: 57–60.
Echeverría C, Montorfano I, Sarmiento D, Becerra A, Nuñez-Villena F, Figueroa XF et al. (2013). Lipopolysaccharide induces a fibrotic-like phenotype in endothelial cells. J Cell Mol Med 17: 800–814.
Fateh-Moghadam S, Htun P, Tomandl B, Sander D, Stellos K, Geisler T et al. (2007). Hyperresponsiveness of platelets in ischemic stroke. Thromb Haemost 97: 974–978.
Fiusa MM, Carvalho-Filho MA, Annichino-Bizzacchi JM, De Paula EV (2015). Causes and consequences of coagulation activation in sepsis: an evolutionary medicine perspective. BMC Med 13: 105.
Frelinger AL 3rd, Jakubowski JA, Brooks JK, Carmichael SL, BernyLang MA, Barnard MR et al. (2014). Platelet activation and inhibition in sickle cell disease (pains) study. Platelets 25: 27–35.
Gavins FNE, Li G, Russell J, Perretti M, Granger DN (2011a). Microvascular thrombosis and CD40/CD40L signalling. J Thromb Haemost 9: 574–581. Gavins FNE, Russell J, Senchenkova EL, De Almeida PL, Damazo AS, Esmon CT et al. (2011b). Mechanisms of enhanced thrombus formation in cerebral microvessels of mice expressing hemoglobin-S. Blood 117: 4125–4133.
Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al. (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106.
Jayakumar T, Chen WF, Lu WJ, Chou DS, Hsiao G, Hsu CY (2013). A novel antithrombotic effect of sulforaphane via activation of platelet adenylate cyclase: ex vivo and in vivo studies. J Nutr Biochem 24: 1086–1095.
Jones S, Solomon A, Sanz-Rosa D, Moore C, Holbrook L, Cartwright EJ (2010). The plasma membrane calcium ATPase modulates calcium homeostasis, intracellular signaling events and function in platelets. J Thromb Haemost 8: 2766–2674.
Jung SM, Moroi M (1998). Platelets interact with soluble and insoluble collagens through characteristically different reactions. J Biol Chem 273: 14827–14837.
Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010a). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG (2010b). Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. J Pharmacol Pharmacother 1: 94–99.
Konić-Ristić A, Srdić-Rajić T, Kardum N, Aleksić-Veličković V, Kroon PA, Hollands WJ et al. (2013). Effects of bioactive-rich extracts of pomegranate, persimmon, nettle, dill, kale and Sideritis and isolated bioactives on arachidonic acid induced markers of platelet activation and aggregation. J Sci Food Agric 93: 3581–3587.
Liu Y, Jennings NL, Dart AM, Du X (2012). Standardizing a simpler, more sensitive and accurate tail bleeding assay in mice. World J Exp Med 2: 30–36. Liu D, Liang F, Wang X, Cao J, Qin W, Sun B (2013). Suppressive effect of CORM-2 on LPS-induced platelet activation by glycoprotein mediated HS1 phosphorylation interference. PLoS One 8: e83112.
Lou J, Donati YR, Juillard P, Giroud C, Vesin C, Mili N et al. (1997). Platelets play an important role in TNF-induced microvascular endothelial cell pathology. Am J Pathol 51: 1397–1405.
Lopes Pires ME, Clarke SR, Marcondes S, Gibbins JM (2017). Lipopolysaccharide potentiates platelet responses via toll-like receptor 4-stimulated Akt-Erk-PLA2 signalling. PLoS One 12: e0186981.
McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M et al. (2016). Heart disease and stroke statistics—2015 update: a report from the American Heart Association. Circulation 133: 447–454.
Patel KN, Soubra SH, Bellera RV, Dong JF, McMullen CA, Burns AR (2008). Differential role of von Willebrand factor and P-selectin on microvascular thrombosis in endotoxemia. Arterioscler Thromb Vasc Biol 28: 2225–2230.
Sakr Y, Dubois MJ, De Backer D, Creteur J, Vincent JL (2004). Persistent microcirculatory alterations are associated with organ failure and death in patients with septic shock. Crit Care Med 32: 1825–1831.
Secor D, Li F, Ellis CG, Sharpe MD, Gross PL, Wilson JX et al. (2010). Impaired microvascular perfusion in sepsis requires activated coagulation and P-selectin-mediated platelet adhesion in capillaries. Intensive Care Med 36: 1928–1934.
Tang YH, Vital S, Russell J, Seifert H, Senchenkova E, Granger DN (2014). Transient ischemia elicits a sustained enhancement of thrombus development in the cerebral microvasculature: Effects of anti-thrombotic therapy. Exp Neurol 261: 417–423.
Tymvios C, Jones S, Moore C, Pitchford SC, Page CP, Emerson M (2008). Real-time measurement of non-lethal platelet thromboembolic responses in the anaesthetized mouse. Thromb Haemost 99: 435–440.
Woth G, Varga A, Ghosh S, Krupp M, Kiss T, Bogár L et al. (2011). Platelet aggregation in severe sepsis. J Thromb Thrombolysis 31: 6–12.
Yan SL, Russell J, Harris NR, Senchenkova EY, Yildirim A, Granger DN (2013). Platelet abnormalities during colonic inflammation. Inflamm Bowel Dis 19: 1245–1253.
Youn HS, Kim YS, Park ZY, Kim SY, Choi NY, Joung SM (2010). Sulforaphane suppresses oligomerization of TLR4 in a thioldependent manner. J Immunol 184: 411–419.
Zhang Y, Talalay P, Cho CG, Posner GH (1992). A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. PNAS 89: 2399–2403. Zhang G, Han J, Welch EJ, Ye RD, Voyno-Yasenetskaya TA, Malik AB (2009). Lipopolysaccharide stimulates platelet secretion and potentiates platelet aggregation via TLR4/MyD88 and the cGMPdependent protein kinase pathway. J Immunol 182: 7997–8004.
Supporting Information
Additional supporting information may be found online in the Supporting Information section at the end of the article. https://doi.org/10.1111/bph.14368 Figure S1 Effect of LPS on velocity of human platelet aggregates. Velocity of aggregate formation was measured using low angle light scattering technique for platelets stimulated by LPS (1 and 7.5 μg·mL1 ) for 5 min or vehicle (saline) treatment. Data are mean ± SEM of n = 3–5 mice/group (0 = 4 mice/group (no response); 1 μg/ml of LPS (n = 3 mice/group. No response) and 7.5 μg·mL1 of LPS (n = 5 mice/group, due to slight, but non-significant response).
Les artikkelen her
bottom of page